Chẩn đoán sớm bệnh Thalassemia để nâng cao chất lượng cuộc sống

Chẩn đoán sớm bệnh Thalassemia để nâng cao chất lượng cuộc sống
1. Bệnh Thalassemia
Thalassemia là bệnh thiếu máu tan máu di truyền, do giảm hoặc mất hẳn sự tổng hợp của một loại chuỗi globin. Tuỳ theo sự thiếu hụt tổng hợp ở chuỗi alpha (α), beta (β), hay cả ở chuỗi delta (δ) và beta mà có tên gọi là α thalassemia, β thalassemia, hay δβ thalassemia
Bệnh gây ra những ảnh hưởng sâu sắc tới sự tăng trưởng, phát triển thể chất và chất lượng cuộc sống của bệnh nhân, giảm khả năng lao động, học tập tạo ra gánh nặng kinh tế cho gia đình và xã hội, thậm chí ảnh hưởng lớn tới sự phát triển nòi giống.

Trước những năm 1970 bệnh thalassemia nặng thường chết trước tuổi trưởng thành, nhiều người bệnh có những di chứng nặng nề. Hiện nay, phương pháp điều trị chủ yếu là truyền máu định kỳ kết hợp với thải sắt và các thuốc hỗ trợ khác đã cải thiện chất lượng cuộc sống cho bệnh nhân thalassemia, nhưng việc xét nghiệm phát hiện bệnh, tư vấn cho người bệnh là vô cùng quan trọng.
2. Lịch sử và dịch tễ
Từ Thalassemia là từ gốc Hy lạp có nghĩa là “Bệnh thiếu máu vùng biển”, do bệnh được phát hiện đầu tiên và phổ biến ở vùng Địa Trung Hải. Năm 1925 Thomas Cooley, một bác sĩ nhi khoa người Mỹ lần đầu tiên miêu tả đặc điểm bệnh nhân trên những bệnh nhi và vậy còn có tên gọi khác là “bệnh thiếu máu Cooley”
Thalassemia phân bố khắp toàn cầu nhưng có tính địa dư rõ rệt: tỷ lệ cao ở Địa Trung Hải,Trung Đông, Châu Á, Thái Bình Dương. Theo số liệu thống kê của WHO, bệnh huyết sắc tố ảnh hưởng tới 71% các nước trên thế giới. Mỗi năm có khoảng 60.000-70.000 trẻ em sinh ra bị bệnh β-thalassemia mức độ nặng.
3. phân loại thalassemia
3.1. α thalassemia
Có 4 gen điều khiển tổng hợp chuỗi α globin nằm trên nhiễm sắc thể 16. Nếu gen gen đó tổn thương do mất đoạn NST hoặc đột biến điểm, thay thế, thêm hoặc mất một vài base nitơ sẽ làm giảm hoặc mất hẳn tổng hợp chuỗi α.
Tùy theo mức độ tổn thương gen mà chia thành các thể bệnh như sau:
Thể α₁thal: Mất một trong bốn gen α.
Thể α₂thal: Mất hai trong bốn gen α.
Thể HbH: Mất ba trong bốn gen α.
Thể Hb Bart’s: Mất cả bốn gen α.
Hb Constant Spring (CS): Mạch α bất thường với số lượng nhỏ như thiếu gen.
Có thể xảy ra sự kết hợp αthal với các Hb bất thường khác về cấu trúc ở cả chuỗi α và β, như HbQ, HbG Philadelphia, HbE, HbJ.
Có thể xảy ra sự kết hợp αthal với các Hb bất thường khác về cấu trúc ở cả chuỗi α và β, như HbQ, HbG Philadelphia, HbE, HbJ.
3.2. β thalassemia
Có 2 gen chi đạo tổng hợp chuỗi β (gen β) nằm trên cánh ngắn nhiễm sắc thể 11 cùng các gen δ, γ, ε. Nếu nhiễm sắc thể tổn thương mất hoàn toàn khả năng chỉ đạo tổng hợp chuỗi β gọi là β⁰. Nếu gen β tổn thương làm giảm tốc độ tổng hợp chuỗi β gọi là β⁺.
Các đột biến ở vùng khởi động làm giảm tốc độ sao chép, làm giảm tổng hợp chuỗi β, gây β⁺ thal.
Đột biến ở một số bộ 3 mã hóa làm thành mã chấm hết không tạo ARNm đầy đủ nên không tổng hợp được chuỗi β, gây β⁰ thal.
Các đột biến ở đoạn đầu hay đoạn cuối sao chép làm rối loạn quá trình sao chép ARNm, gây giảm tổng hợp chuỗi β gây β⁺ thal.
Các đột biến ở vùng intron làm chậm quá trình chỉnh sửa của ARNm gây β⁺ thal.
Các dạng tổn thương khác của chuỗi β:
Không tổng hợp được chuỗi δ và γ, ít có ý nghĩa lâm sàng.
Các thể phối hợp hay gặp:
β thal/ Hb E: biểu hiện lâm sàng nặng giống như β thal đồng hợp tử.
β thal/ Hb S.
β thal/ Hb C.
4. Biến chứng
4.1. Quá tải sắt (chiếm chủ yếu)
Khi quá tải sắt, độ bão hòa sắt cao trên 50%, sắt sẽ gắn không đặc hiệu với các chất khác như albumin, citrate, aminoacid và đường. Những ion sắt gắn không đặc hiệu này dễ dàng bị thay đổi trạng thái từ Fe³⁺ thành Fe²⁺ sinh ra các ion hình thành các gốc tự do, gây tổn thương các phân tử như màng lipid, hạt trong tế bào và DNA, hậu quả làm tế bào chết và hình thành tổ chức sợi.
Các tổ chức bị tổn thương do quá tải sắt là:
- Tuyến nội tiết (Suy tuyến yên, suy tuyến giáp, suy cận giáp, suy tuyến tụy, suy tuyến sinh dục)
- Tim: Do quá trình thiếu máu kéo dài làm tim phải tăng cường hoạt động và cơ tim bị nhiễm sắt do ứ đọng sắt dẫn đến rối loạn nhịp tim, cơ tim xơ hóa, suy tim.
Biến chứng tại tim là nguyên nhân chính gây ra tử vong cho bệnh nhân Thalassemia, với thalassemia mức độ trung bình biến chứng tim thường xảy ra khoảng 10 năm sau khi bắt đầu được truyền máu mà không có thải sắt.
- Gan: Xơ gan là hậu quả của nhiễm sắt và có thể do nhiễm virus viêm gan do truyền máu nhiều lần.
- Da: Xạm da do lắng đọng sẳt, lở loét chân do tuần hoàn vi mạch kém
4.2 Xương, khớp: Loãng xương và phì đại xương dẹt. Nguyên nhân:
Do tổ chức sinh máu trong xương dẹt quá sản để tăng tạo máu làm phì đại xương đầu, mặt.
Thiếu hormon tăng trưởng, hormon sinh dục làm chậm quá trình phát triển của xương.
4.3. Huyết khối.
Giả thiết cơ chế gây huyết khối:
Cắt lách: làm tăng β2 thromboglobulin, tăng số lượng hồng cầu bất thường, tăng số lượng tiểu cầu.
Huyết khối có thể xảy ra tại: có thể xảy ra ở não, tĩnh mạch, phổi
5. Xét nghiệm chẩn đoán bệnh.
5.1. Tổng phân tích tế bào máu bào máu ngoại vi
- Hồng cầu nhỏ đặc trưng cho bệnh thalassemia
- Số lượng hồng cầu, HGB, HCT,MCV, MCH là các chỉ số đặc biệt quan trọng
5.2. Huyết đồ
- Hồng cầu nhỏ
- Hồng cầu đa hình thái :Hồng cầu hình giọt nước, Hồng cầu hình bia, Hồng cầu hình liềm , Hồng cầu hình bầu dục, Hồng cầu hình cầu, Hồng cầu hình gai
- Mảnh vỡ hồng cầu
5.3 Hồng cầu lưới : số lượng hồng cầu lưới tăng
5.4. Tủy đồ
- Xét nghiệm tủy đồ có ý nghĩa quan trọng trong chẩn đoán thalassemia, đặc biệt là trong việc đánh giá tình trạng tăng sinh của dòng hồng cầu và xác định các bất thường trong quá trình tạo máu.
-Tủy đồ giúp xác định nguyên nhân của thiếu máu ở bệnh nhân thalassemia, loại trừ các nguyên nhân khác như thiếu sắt hoặc thiếu máu do các bệnh lý khác.
-Tủy đồ cũng có thể được sử dụng để theo dõi đáp ứng của bệnh nhân với các phương pháp điều trị như truyền máu hoặc điều trị bằng thuốc.
5.5. Sức bền thẩm thấu hồng cầu: Độ thẩm thấu hồng cầu tăng
5.6.Điện di huyết sắc tố: có huyết sắc tố bất thường.
5.7. PCR: xác định đột biến gen.
5.8. Kết hợp với 1 số xét nghiệm hóa sinh: Nồng độ ferritin huyết thanh, sứt huyết thanh
6. Phòng bệnh
Tư vấn di truyền và chẩn đoán trước sinh
- Tư vấn di truyền : những đôi trai gái muốn kết hôn mà trong gia đình có người mắc bệnh nên kiểm tra
+ Xét nghiệm Tổng phân tích tế bào máu bào máu ngoại vi
+ Điện di huyết sắc tố
+ Sức bền thẩm thấu hồng cầu
+ Không kết hôn giữa 2 người mang gen β-thalassemia thể nhẹ hoặc thalassemia với HbE, α với α thalassemia và hướng dẫn chẩn đoán trước sinh kết hợp tư vấn dịch vụ sản khoa và xét nghiệm ADN
- Sàng lọc cộng đồng để phát hiện người mang gen Thalassemia và tư vấn di truyền cho các cặp vợ chồng có nguy cơ là những biện pháp cốt lõi và hiệu quả nhất để phòng ngừa sinh con mắc các thể Thalassemia nặng, qua đó giảm gánh nặng bệnh tật cho gia đình và xã hội. Việc này đặc biệt quan trọng ở các quốc gia có tỷ lệ người mang gen cao như Việt Nam.
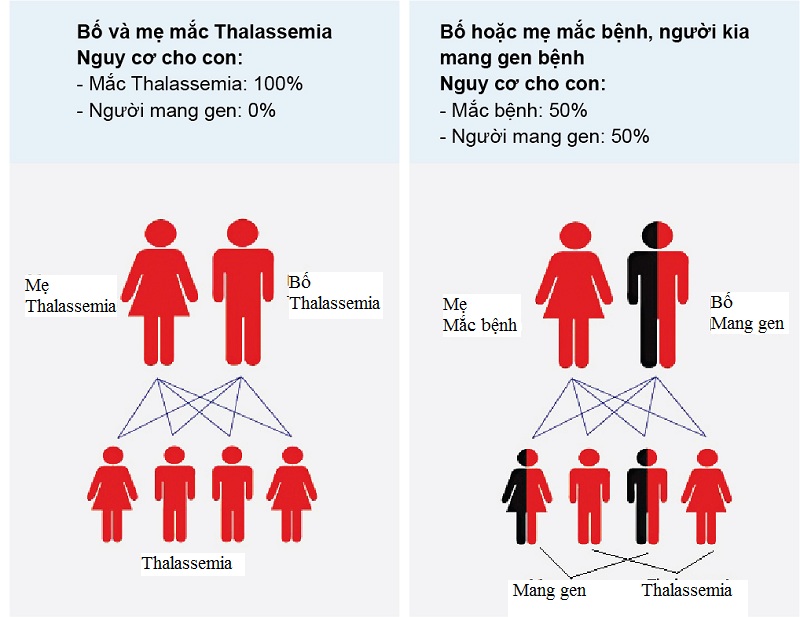
Tỷ lệ con sinh ra mắc Thalassemia khi bố hoặc mẹ mắc Thalassemia
Nếu cần tư vấn về khám sàng lọc bệnh Thalassemia, hãy liên hệ với chuyên gia Bệnh viện đa khoa Hà Đông. Với đội ngũ chuyên gia, bác sỹ giàu kinh nghiệm, hệ thống máy xét nghiệm hiện đại, BV ĐK Hà Đông là địa chỉ tin cậy để bạn gửi gắm niềm tin.
Liên hệ tổng đài chăm sóc khách hàng 1900866689 hoặc 02433528203 để được tư vấn chi tiết các dịch vụ.
Hình ảnh chuyên gia và hệ thống máy tại Khoa Huyết học Truyền máu./.
Hệ thống máy tổng phân tích tế bào máu ngoại vi Advia 2120i

Máy Điện di Huyết sắc tố
Tài liệu tham khảo
1.Nguyễn Thị Thu Hà, Ngô Mạnh Quân (2014), “Khảo sát hiểu biết, thái độvà thực hành về bệnh tan máu bẩm sinh của bốmẹtrẻmắc tan máu bẩm sinh tại viện Huyết học –Truyền máu Trung ương”.Tạp chí Y học thực hành.
2. Mostafa S, Elaziz M.A(2014). Factors Affecting Compliance Plan of Thalassemic Children and their Mothers in Outpatient Clinic at Zagazig University Hospitals. Journal of Biology, Agriculture and Healthcare, Vol.4, No.3
3.Bộ Y tế, Viện Huyết học truyền máu trung ương 1 số chuyên đè huyết học truyền máu, tập 3, chủ biên PSG,TS nguyễn nah trí (2010)
4.Bệnh huyết sắc tố, tác giả Teruo Harano, dịch giả Ths, Bs Bạch Quốc Khánh (2010).
Tham vấn chuyên môn
CKI- Đặng Thị Hảo
Cùng xem